A proteína CFTR e seus moduladores | Entrevista com Dr. Miquéias Lopes Pacheco
Instituto Unidos pela Vida - 26/04/2017 17:00
Miquéias Lopes Pacheco
Falamos com o biomédico, Dr. Miquéias Lopes Pacheco, no VI Congresso Brasileiro de Fibrose Cística. Miquéias faz pós-doutorado em Portugal sobre Fibrose Cística (FC), é irmão de um paciente e busca junto a outros pesquisadores encontrar os melhores tratamentos possíveis para pessoas com FC, independente de suas mutações. Ele nos explica aqui o que é CFTR, como ele atua no organismo de uma pessoa com FC e como agem os seus moduladores. Confira:
Unidos pela Vida: O que é CFTR?
Dr. Miquéias: É um gene que está presente no cromossomo 7 do genoma humano. Ele codifica uma proteína que também é chamada de CFTR. Quando essa proteína é uma proteína normal, ela está presente na membrana plasmática (aquele envolto que delimita uma célula) em diversos tecidos do nosso corpo, como por exemplo nas glândulas sudoríparas, no pâncreas, nas vias aéreas e no canal deferente, no caso dos homens.
Unidos pela Vida: Como o CFTR age no organismo?
Dr. Miquéias: O CFTR atua como um canal de cloreto, ou seja, ele forma um poro na membrana das células para que ocorra a passagem de cloreto. Isso permite que haja um equilíbrio entre a quantidade de água dentro e fora das células, mantendo um revestimento líquido sobre as células (como se fosse uma “capa protetora”). Recentemente também foi descoberto que o CFTR atua como um transportador de bicarbonato. Esse é um fator importante porque o bicarbonato está envolvido com a eliminação de patógenos (bactérias e fungos). Por essa razão que pacientes com Fibrose Cística tem dificuldades de eliminar patógenos nos pulmões, o que leva a infecções recorrentes.
Unidos pela Vida: O que são as mutações residuais? Qual a diferença entre essas mutações e as demais mutações presentes no CFTR?
Dr. Miquéias: Existem diversas mutações no CFTR. No banco de dados conhecido como CFTR1 existem mais de 2 mil mutações reportadas e uma grande quantidade delas causam a Fibrose Cística, quando há mutação nos dois alelos. O que nós estamos tentando fazer, atualmente, é entender como cada uma destas mutações afeta a proteína CFTR. Dessa forma, podemos dividir essas mutações em grupos de acordo com o defeito que elas causam na proteína e buscar tratamentos mais adequados para corrigir estes defeitos (o que nós chamamos de terapias personalizadas ou medicina de precisão). Como por exemplo, existem mutações em que a proteína é codificada, mas ela é rapidamente degradada (chamadas de classe II, ex.: F508del). Também existem mutações em que a proteína é codificada, chega na membrana plasmática das células, mas a proteína não funciona e, por isso, não ocorre passagem de cloreto (chamadas de classe III, ex.: G551D). As mutações residuais são mutações de classe IV. Nestes casos, a proteína CFTR é codificada, chega na membrana plasmática das células, mas ela tem a sua função reduzida comparada ao CFTR normal. Um exemplo de mutação de classe IV é a R117H, uma das mais comuns. Em teoria, uma pessoa que tem uma mutação residual apresenta um fenótipo (ou severidade) mais brando da doença, não sendo um quadro tão grave.
Unidos pela Vida: O que são e para que servem os moduladores do CFTR? 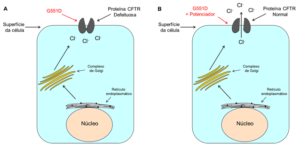
Dr. Miquéias: São essas novas drogas que estamos tentando desenvolver e que irão atuar de acordo com defeitos específicos causados pelas mutações. Atualmente, os tratamentos se baseiam em mucolíticos, antibióticos, enzimas pancreáticas, entre outros (os quais são muito importantes!). Contudo, eles atuam numa fase mais tardia da cascata patológica da doença. E o que nós queremos é encontrar tratamentos quem atuem numa fase mais precoce desta cascata. Ou seja, atuar diretamente na causa da doença.
O primeiro modulador do CFTR aprovado para tratamento dos pacientes foi o Kalydeco (substância ativa: Ivacaftor). O Kalydeco pode atuar em mutações de classe III e IV. Como eu havia mencionado anteriormente, nestas mutações a proteína CFTR é codificada, ela chega na membrana plasmática das células, só que ela não funciona ou funciona bem menos do que o normal. O que o Kalydeco faz é interagir com este CFTR, permitindo que a proteína volte a funcionar (ou seja, permitir a passagem de cloreto).
Mais recentemente foi aprovado o tratamento dos pacientes homozigotos para a mutação F508del com outro modulado, o Orkambi (combinação das substâncias ativas: Lumacaftor e Ivacaftor). O Lumacaftor pode atuar em mutações de classe II, no qual a proteína CFTR é codificada, mas não consegue enovelar (ou dobrar) totalmente e por isso é degradada. Podemos fazer uma analogia ao origami, no qual você tem um papel e todas aquelas dobraduras que devem ser criteriosamente seguidas para conseguir obter o formato final. O Lumacaftor vai ajudar o CFTR para que a proteína atinja esta estrutura enovelada e, posteriormente, chegue à
membrana plasmática das células.
Unidos pela Vida: Você está fazendo pós-doutorado em Portugal sobre Fibrose Cística. Sobre o que é a sua pesquisa e qual o objetivo dela?
Dr. Miquéias: Um dos objetivos do meu trabalho é encontrar fármacos cada vez melhores e que trarão maiores efeitos benéficos aos pacientes com Fibrose Cística. Para isso, é importante entender como estes fármacos atuam (saber qual o mecanismo de ação subjacente ao tratamento). Além disso, sabemos que existem muitas mutações raras no CFTR (também chamadas de mutações órfãs). Nós estamos analisando qual o impacto destas mutações raras sobre a estrutura do CFTR para que com essa melhor caracterização possamos buscar fármacos que beneficiariam estes pacientes.
Unidos pela Vida: Que mensagem você gostaria de deixar para as pessoas com FC?
Dr. Miquéias: Muito esforço e dedicação tem sido colocados por diversos pesquisadores de diversas partes do mundo. Acho que a mensagem mais importante e que deve ficar é que nós da área da pesquisa básica, em cooperação aos pesquisadores da área clínica, estamos na árdua busca para encontrar os melhores tratamentos possíveis para os pacientes com Fibrose Cística, sejam eles com mutações comuns ou raras.
Miquéias Lopes Pacheco: é biomédico pela UNIPAC (2009). Mestre (2012) e Doutor (2016) em Ciências Biológicas/Biofísica pelo Instituto de Biofísica Carlos Chagas Filho (IBCCF) da UFRJ. Realizou parte do Doutorado no Departamento de Fisiologia da Johns Hopkins University. Atualmente é Pós-Doutor/Colaborador no Instituto de Biossistemas e Ciências Integrativas (BioISI) da Universidade de Lisboa (FCUL). É do interior de Minas Gerais e tem um irmão de 24 anos com FC, cujo diagnóstico veio tardiamente, aos 6 anos de idade. Ele conta que seu interesse em estudar sobre a Fibrose Cística veio ainda na faculdade. “O meu desejo de trabalhar com FC surgiu no 5º período da faculdade de Biomedicina, na matéria de Genética Básica, na qual cada grupo deveria escolher uma doença genética para apresentar um trabalho. Eu sabia o que era FC, mas não compreendia a fundo a doença. Comecei a estudar e me apaixonei pelo assunto! E falei pra mim mesmo: – São essas pessoas que eu espero, fazendo pesquisa, ajudar! Depois disso acabei indo para o Rio de Janeiro fazer Mestrado e Doutorado em um laboratório que trabalha com CFTR para começar a me envolver no campo”. Relembre o depoimento do Dr. Miquéias, publicado em 2016 aqui em nossa página: http://bit.ly/DepoimentoMiquéias
Agradecemos ao Dr. Miquéias Pacheco pela importante participação e entrevista! =)
===
Nota importante: As informações aqui contidas tem cunho estritamente educacional. Em hipótese alguma pretendem substituir a consulta médica, a realização de exames e ou, o tratamento médico. Em caso de dúvidas fale com seu médico, ele poderá esclarecer todas as suas perguntas.


