Entendendo melhor a Fibrose Cística
Instituto Unidos pela Vida - 15/08/2016 11:53Já ouvimos falar bastante que a Fibrose Cística (FC) é um distúrbio genético autossômico recessivo (transmitido através da família) que pode afetar principalmente os pulmões, pâncreas, fígado e intestino. Os sintomas acontecem porque ocorre no organismo um transporte anormal de cloreto e sódio através da membrana celular, deixando a secreção mais espessa. A FC é uma das doenças raras mais comuns, e tem sintomas que podem ser facilmente confundidos com outras doenças, podendo afetar também as glândulas sudoríparas e o sistema reprodutivo masculino.
Os principais sintomas da FC incluem dificuldade na respiração, a tosse crônica, as infecções do seios paranasais, algumas limitações no crescimento e até infertilidade. É causada por mutações na proteína CFTR (regulador de condutância transmembranar de fibrose cística, na sigla em inglês). Esse canal proteico regula o suor, os fluidos digestivos e o muco. A CFTR controla o movimento dos íons de cloreto e sódio nas membranas celulares como o epitélio dos alvéolos nos pulmões. Geralmente, os indivíduos sem CF têm duas cópias do gene do CFTR. Para o desenvolvimento da FC (pela recessividade), ambas cópias devem conter mutações.
De acordo com o National Heart, Lung and Blood Institute (NHLBI), milhões de indivíduos nos Estados Unidos são portadores do gene defeituoso para CF, apesar da ausência de sintomas. O motivo disso é que o indivíduo com CF deve herdar dois genes defeituosos (um gene de cada pai). Foi estimado que 1 a cada 29 caucasianos (descentendes de europeus) nos Estados Unidos têm o gene para FC. Nos Estados Unidos, a maioria das crianças são diagnosticadas por volta dos 2 anos de idade. Alguns indivíduos com FC não são diagnosticados até os 18 anos ou mais por terem uma forma mais sutil da doença.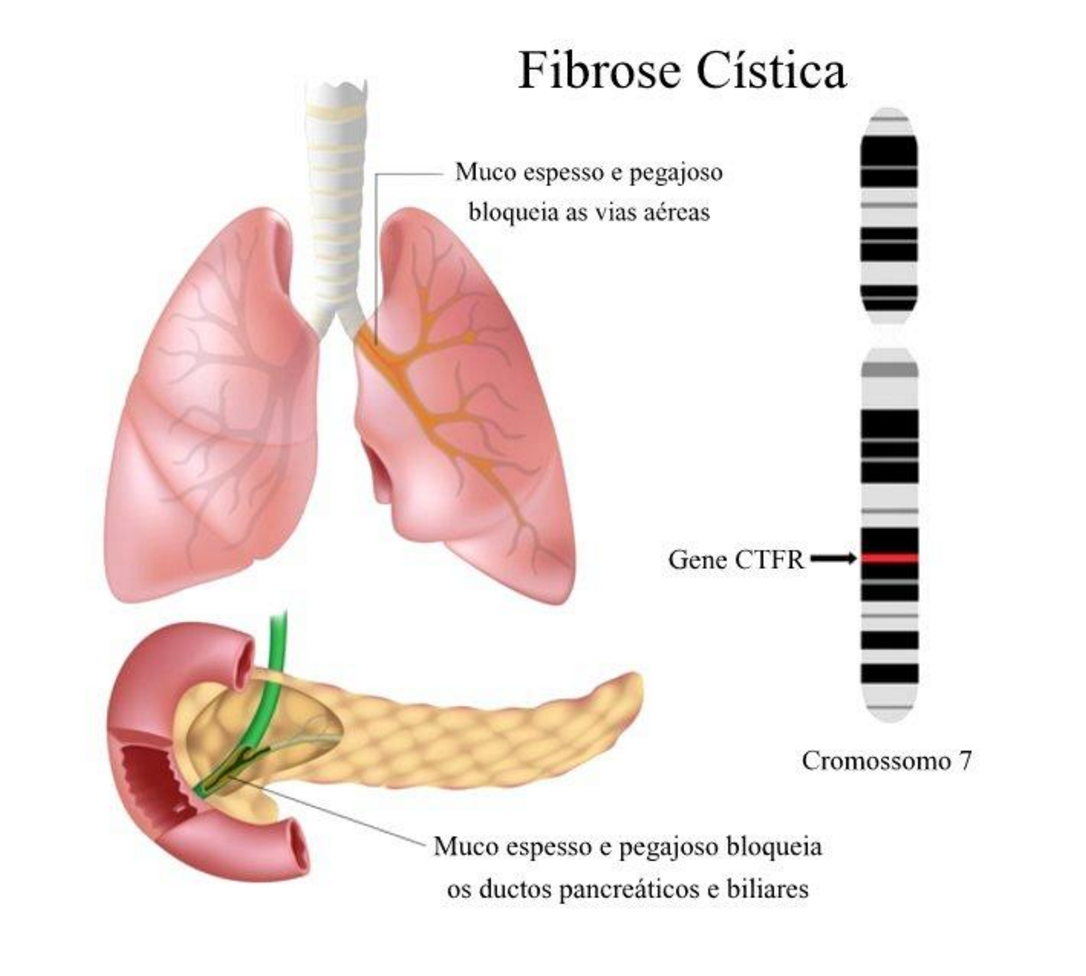
Mecanismo da doença
A proteína do CFTR funciona como um canal entre as membranas celulares e gera o muco, suor, saliva e lágrimas, bem como as enzimas digestivas. Este canal tem a capacidade de transportar íons de cloreto para dentro e para fora de certas células. O transporte de cloreto controla o movimento da água para dentro e fora das células (lembre-se de que a água segue o sal). Isso é importante para a produção de muco que flua livremente, o que lubrifica e protege as vias aéreas, o trato digestivo, o trato reprodutivo, bem como outros tecidos. O CFTR também regula os canais de sódio.
Mutações no gene do CFTR localizados no cromossomo 7 alteram a função do canal de cloreto, impedindo o fluxo normal dos íons de cloreto e da água para dentro e para fora das células. O tecido produz um muco anormalmente espesso que se acumula, afetando pulmões, pâncreas, fígado e trato instestinal.
Até hoje, mais de 2.000 mutações no gene do CFTR foram catalogadas. A maioria dessas mutações alteram um só aminoácido na proteína do CFTR ou deleta uma parte do DNA. A mutação mais comum é conhecida como Delta F508, envolvendo a deleção de uma posição aminoácida na posição 508 na proteína. Essa proteína anormal colapsa depois de ter sido feita, portanto, nunca chega à membrana celular. Dependendo das mutações, isso muda a produção, estrutura ou estabilidade do canal de cloreto. As células que carregam as mutações que alinham as passagens aéreas dos pulmões, pâncreas e outros tecidos produzem um muco anormalmente espesso que bloqueia as vias aéreas e outras glândulas.
Formas da Fibrose Cística
Considerando que existem mais de 2 mil mutações estudadas para a doença, que são divididas em 7 classes, há muitas diferentes formas de FC, variando de manifestações clínicas mais brandas até mais severas.
No entanto, há duas mais notáveis que incluem a mutação Delta F508 e a mutação R117H. A Delta F508 é a mutação mais comum, sendo responsável por cerca de 70% de todos os casos de FC.
Para descobrir qual é a mutação, faz-se necessário a realização de um exame genético que identificará qual é a mutação no gene CFTR.
Fonte:http://cysticfibrosisnewstoday.com/cystic-fibrosis-overview/
Traduzido por: Victor Pereira, voluntário de tradução de textos do Instituto Unidos pela Vida.
Nota importante: As informações aqui contidas têm cunho estritamente educacional. Em hipótese alguma pretendem substituir a consulta médica, a realização de exames e ou, o tratamento médico. Em caso de dúvidas fale com seu médico, ele poderá esclarecer todas as suas perguntas.


